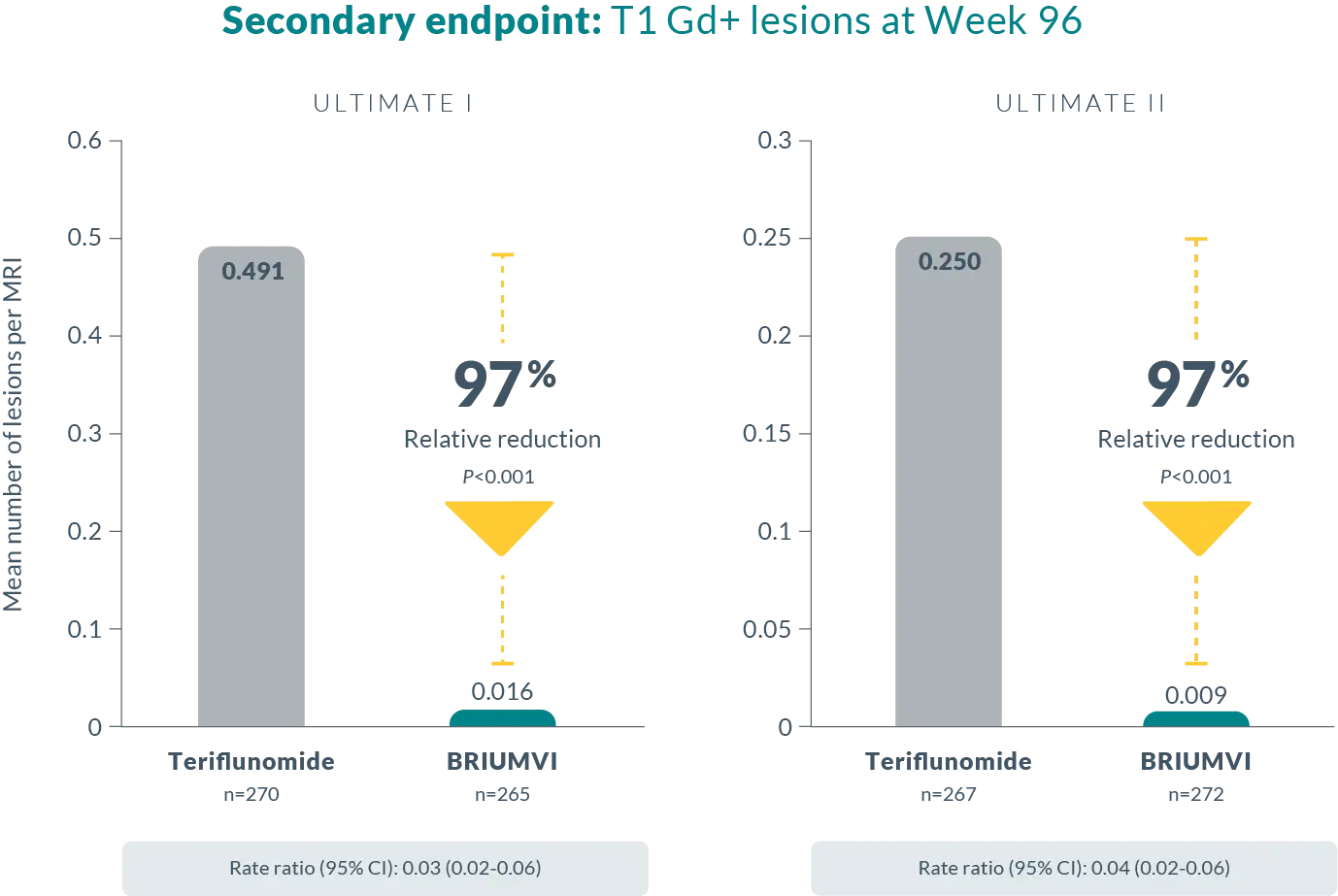
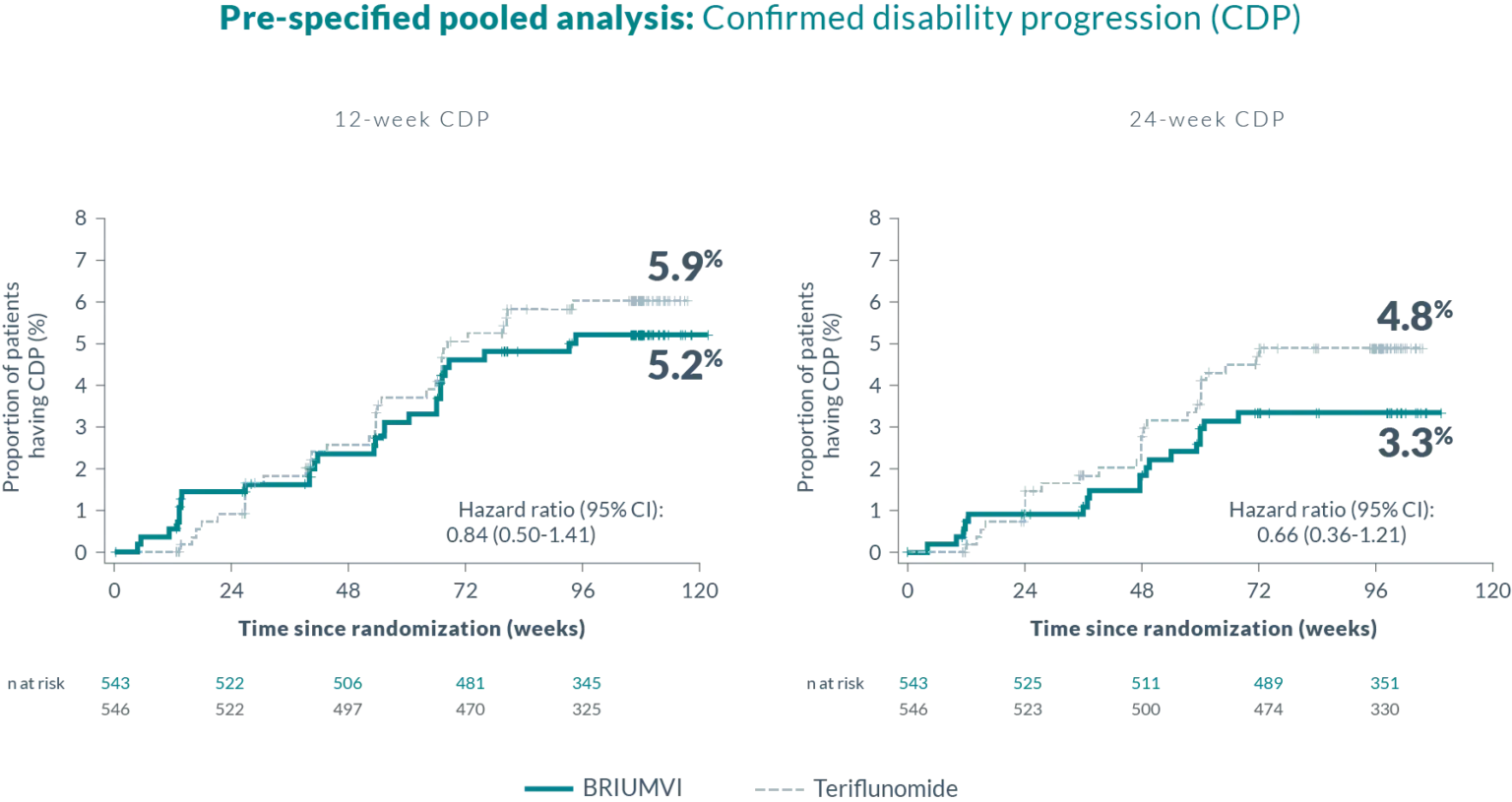
EFFICACY DATA







Indication and Important Safety Information
INDICATION: BRIUMVI is indicated for the treatment of relapsing forms of multiple sclerosis (MS), to include clinically isolated syndrome, relapsing-remitting disease, and active secondary progressive disease, in adults.
IMPORTANT SAFETY INFORMATION: Contraindication: BRIUMVI is contraindicated in patients with:
- Active HBV infection
- A history of life-threatening infusion reaction to BRIUMVI
|
|
These infusion reactions can happen
over 24 hours after your infusion. It is important that
you call your healthcare provider right away if you get any of the
signs or symptoms listed above after each infusion. If you get an
infusion reaction, your healthcare provider may need to stop or slow
down the rate of your infusion.
- Infection:
- Infections are a common side effect, and upper respiratory tract infections are one of the most common side effects of BRIUMVI. BRIUMVI increases your risk of getting infections caused by bacteria or viruses that may be life-threatening or cause death. Tell your healthcare provider if you have an infection or have any of the following signs of infection including fever, chills, a cough that does not go away, or painful urination. Your healthcare provider should delay your treatment with BRIUMVI until your infection is gone.
- Hepatitis B virus (HBV) reactivation: Before starting treatment with BRIUMVI, your healthcare provider will do blood tests to check for hepatitis B viral infection. If you have ever had hepatitis B virus infection, the hepatitis B virus may become active again during or after treatment with BRIUMVI. Hepatitis B virus becoming active again (called reactivation) may cause serious liver problems including liver failure or death. Your healthcare provider will monitor you if you are at risk for hepatitis B virus reactivation during treatment and after you stop receiving BRIUMVI.
- Weakened immune system: BRIUMVI taken before or after other medicines that weaken the immune system could increase your risk of getting infections.
- Progressive Multifocal Leukoencephalopathy (PML): PML may happen with BRIUMVI. PML is a rare, serious brain infection caused by a virus that may get worse over days or weeks. PML can result in death or severe disability. Tell your healthcare provider right away if you have any new or worsening neurologic signs or symptoms. These symptoms may include weakness on one side of your body, loss of coordination in arms and legs, vision problems, changes in thinking and memory which may lead to confusion, and personality changes.
- Low immunoglobulins: BRIUMVI may cause a decrease in some types of antibodies. Your healthcare provider will do blood tests to check your blood immunoglobulin levels.
Before receiving BRIUMVI, tell your healthcare provider about all of your medical conditions, including if you:
- have or think you have an infection.
- take or plan to take medicines that affect your immune system. These medicines may increase your risk of getting an infection.
- have ever had hepatitis B or are a carrier of the hepatitis B virus.
- have had
a recent vaccination or are
scheduled to receive any vaccinations.
- You should receive any required ‘live’ or ‘live-attenuated’ vaccines at least 4 weeks before you start treatment with BRIUMVI. You should not receive ‘live’ or ‘live-attenuated’ vaccines while you are being treated with BRIUMVI and until your healthcare provider tells you that your immune system is no longer weakened.
- When possible, you should receive any ‘non-live’ vaccines at least 2 weeks before you start treatment with BRIUMVI. If you would like to receive any non-live vaccines while you are being treated with BRIUMVI, talk to your healthcare provider.
- If you have a baby and you received BRIUMVI during your pregnancy, it is important to tell your baby’s healthcare provider about receiving BRIUMVI so they can decide when your baby should be vaccinated.
- are pregnant, think that you might be pregnant, or plan to become pregnant. BRIUMVI may harm your unborn baby. You should use birth control (contraception) during treatment with BRIUMVI and for at least 6 months after your last infusion of BRIUMVI. Talk with your healthcare provider about what birth control method is right for you during this time.
- are breastfeeding or plan to breastfeed. It is not known if BRIUMVI passes into your breast milk. Talk to your healthcare provider about the best way to feed your baby if you take BRIUMVI.
Tell your healthcare provider about all the medicines you take, including prescription and over-the-counter medicines, vitamins, and herbal supplements.
What are the possible side effects of BRIUMVI?
The most common side effects of
BRIUMVI include:
- Infusion reactions, upper and lower respiratory tract infections, herpes infections, extremity pain, insomnia, and fatigue.
These are not all the possible side effects of BRIUMVI. Call your doctor for medical advice about side effects. You may report side effects to FDA at 1-800-FDA-1088. You may also report side effects to TG Therapeutics at 1-877-TGTXINC (1-877-848-9462).
For more important information, go to www.briumvi.com or call 1-833-BRIUMVI (1-833-274-8684).